Sugar and alcohol may seem unrelated, but they share multiple “temptation” pathways—from neural reward mechanisms to liver metabolism. Epidemiological studies show that heavy drinkers often have a stronger preference for sugar, and in animal models, alcohol consumption and sugar-seeking behaviors reinforce each other. Their offspring are also more likely to favor high-sugar diets. This phenomenon stems from a common metabolic origin: the fructose pathway. Fructose not only contributes to energy storage and fat synthesis but also activates the dopamine system, triggering “reward pleasure.” Alcohol use disorder (AUD) and alcohol-related liver disease (ALD) are major global public health challenges, with ALD progressing to cirrhosis and high mortality. Previous research indicated that alcohol upregulates aldose reductase (AR) to activate the polyol pathway, generating endogenous fructose from glucose. Ketohexokinase (KHK), the key enzyme in fructose metabolism, plays a pathological role in fatty liver and metabolic hepatitis. This study, published in Nature Metabolism by researchers from the University of Colorado School of Medicine, systematically demonstrates that: alcohol intake drives drinking behavior and liver injury through activation of endogenous fructose metabolism, with KHK as a critical mediator.
Alcohol Preference Depends on Fructose Metabolism
Researchers first observed in chronic alcohol-consuming mice that KHK-A/C isoform mRNA levels and enzyme activity were significantly upregulated in the liver, with ethanol primarily activating the C isoform (KHK-C), not A. In the “two-bottle choice” paradigm, KHK-A/C knockout mice showed a marked reduction in alcohol preference and intake, consistent across sexes and sustained over 30 weeks. In contrast, mice with only KHK-A knockout behaved like wild-type controls, confirming the central role of KHK-C in alcohol preference. Conditioned place preference (CPP) tests further showed that wild-type mice spent significantly more time in the “alcohol-paired chamber,” while KHK-A/C knockout mice exhibited almost no preference. Behaviorally, KHK-deficient mice displayed altered locomotor responses to ethanol—initial brief excitation followed by rapid attenuation—indicating impaired reward signaling. These findings confirm that the fructose metabolism pathway contributes to reward circuit formation.
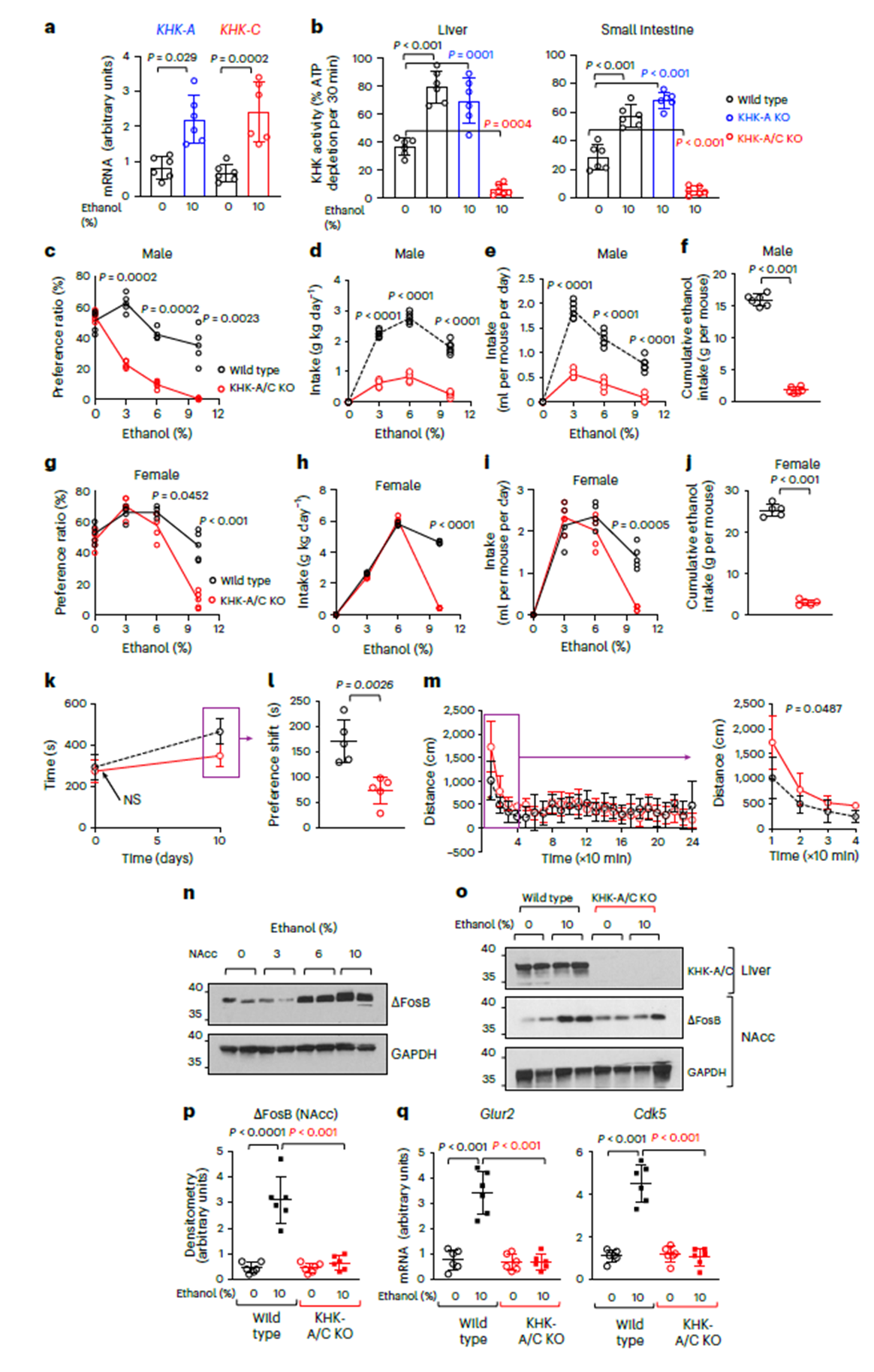
Figure 1. Endogenous fructose metabolism via KHK enhances ethanol intake and preference in mice
Ethanol Induces Endogenous Fructose Synthesis
Since drinking behavior depends on KHK, can ethanol itself trigger fructose metabolism? The study found that ethanol does not increase intestinal fructose transporter GLUT5 expression but instead activates aldose reductase (AR) via osmotic stress. Within 15 minutes of oral ethanol administration, portal vein osmolality rose significantly and correlated with ethanol concentration. Diluting ethanol to 5% nearly abolished this effect. Protein analysis showed that four days of gavage with concentrated ethanol (25%) significantly induced hepatic AR upregulation, while the diluted group showed no change. Subsequent assays confirmed concurrent increases in liver sorbitol and fructose levels, establishing ethanol’s activation of the polyol pathway through hyperosmotic stress. To validate behavioral relevance, AR knockout mice were tested. AR knockout mice exhibited significantly reduced alcohol intake and preference, inversely proportional to AR gene dosage. This links the metabolic chain: ethanol → AR → fructose → KHK, revealing the endogenous source of alcohol’s reinforcing effects.
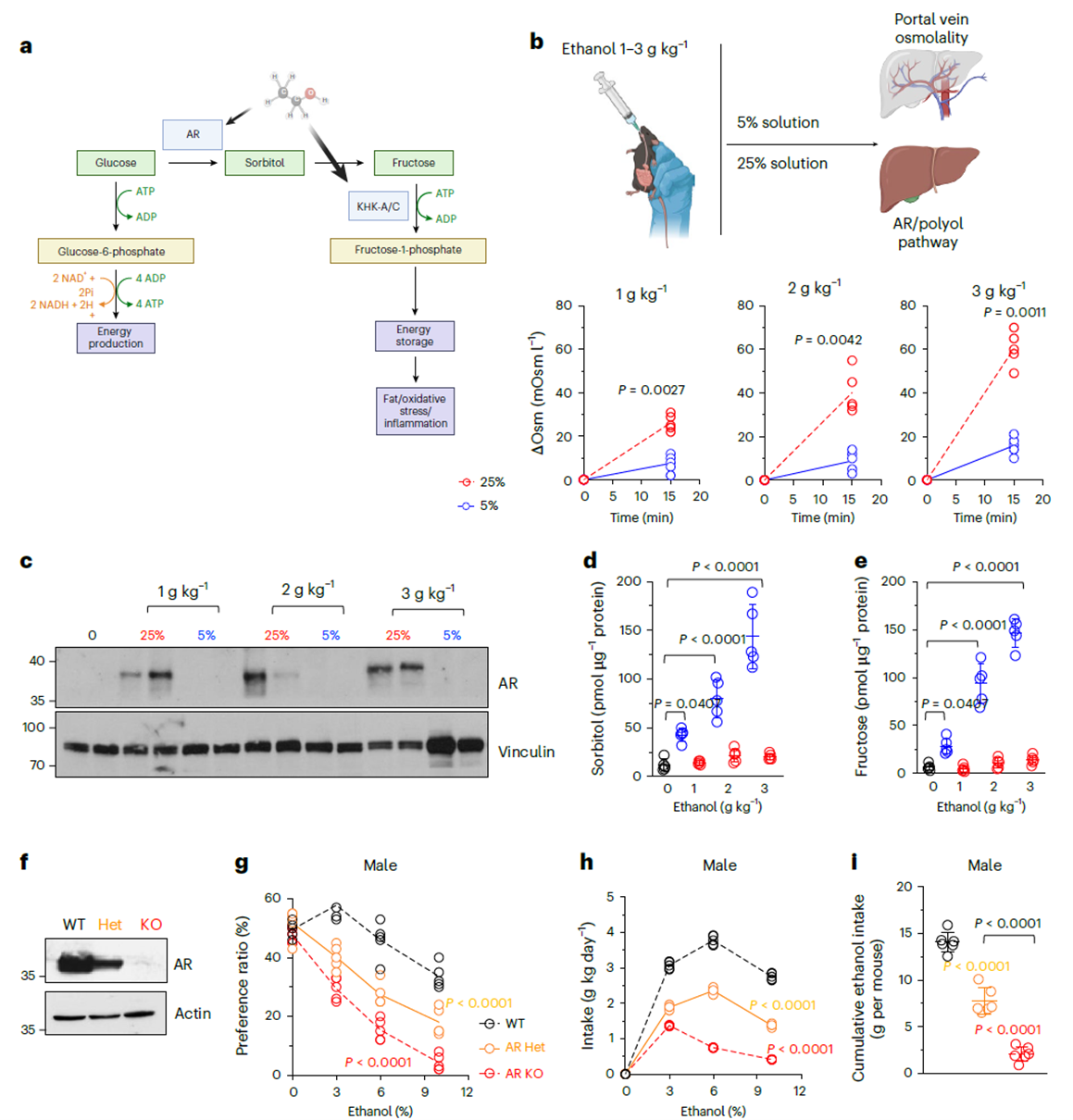
Figure 2. Ethanol-mediated endogenous fructose production and phenotypic response
Dual Liver–Intestine Regulation: Fructose Metabolism Influences Behavior and Metabolism
To dissect tissue-specific contributions, the team generated liver-specific (Alb1-cre) and intestine-specific (Vil1-cre) KHK-A/C knockout models.
Liver-specific KHK knockout mice (Khkfl/fl×Alb1-cre) showed significantly reduced alcohol preference and impaired acetaldehyde metabolism. Following ethanol exposure, ALDH1A1 and ALDH2 expression dropped markedly, leading to elevated plasma acetaldehyde and suppressed central reward signaling (∆FosB/Glur2/Cdk5). Thus, liver fructose metabolism regulates both ethanol detoxification and reward sensitivity.
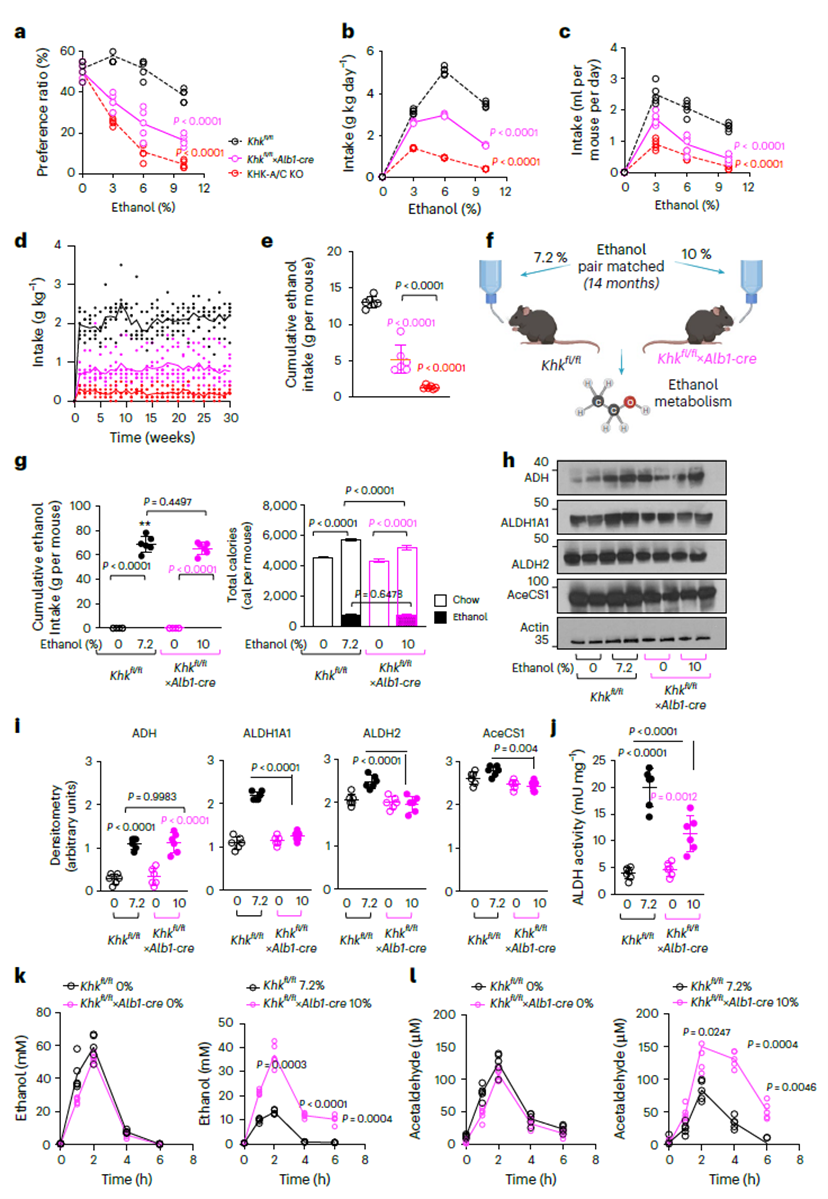
Figure 3. Liver-derived factors linked to endogenous fructose metabolism drive ethanol intake, preference, and metabolism
Intestine-specific KHK knockout mice (Khkfl/fl×Vil1-cre) also drank less, but via a different mechanism. Ethanol induced AR and KHK upregulation in the small intestine and fructose accumulation. It also suppressed GLP-1 secretion, which was restored by the KHK inhibitor CRP427 (Fig. 4g–k). In vivo, ethanol significantly reduced plasma GLP-1, an effect attenuated in KHK knockout models (Fig. 4l). Thus, intestinal KHK modulates drinking motivation via the GLP-1 signaling axis, forming a “central–peripheral” dual-pathway network with liver metabolism.
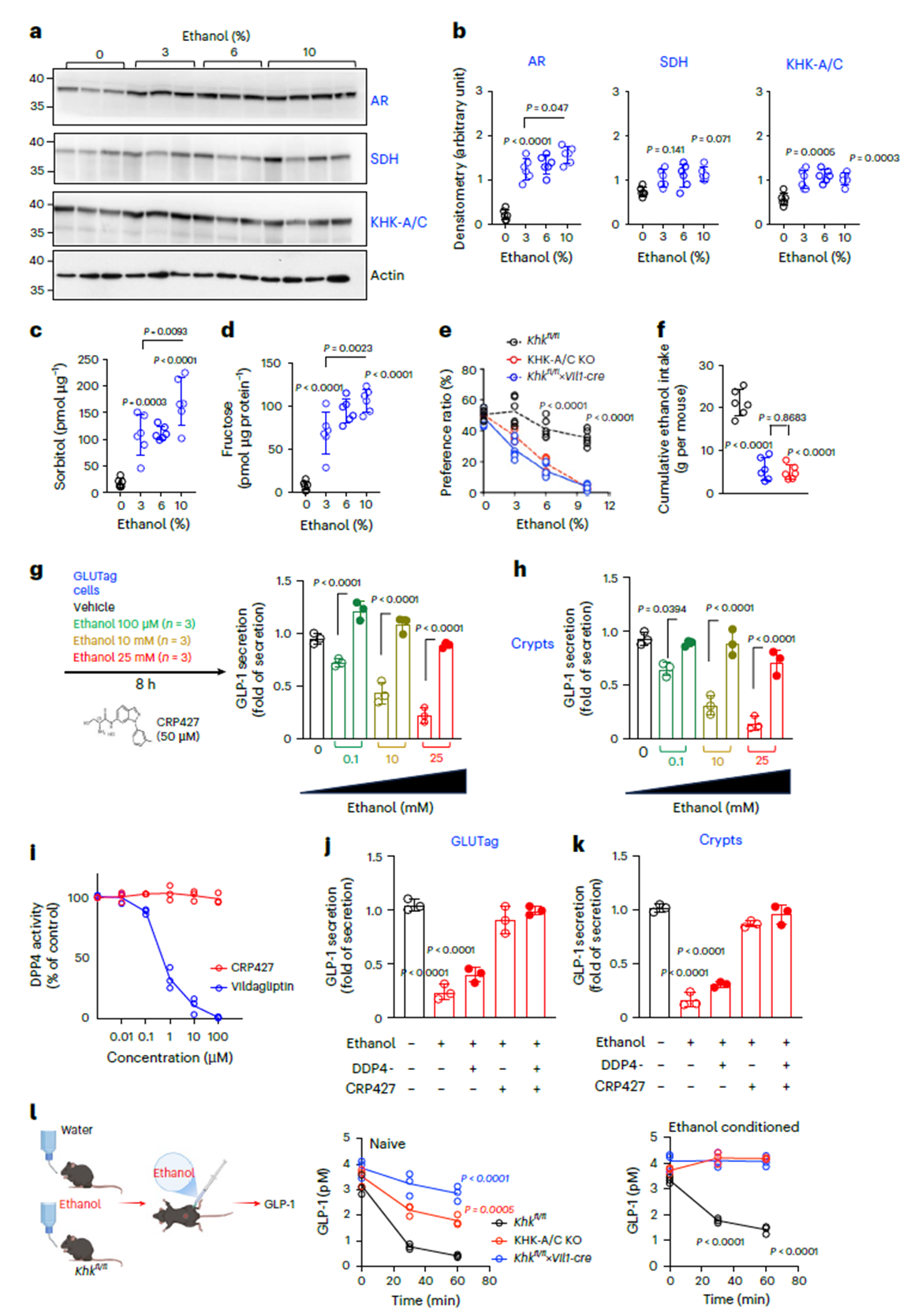
Figure 4. Intestine-derived factors linked to endogenous fructose metabolism drive ethanol intake and preference
Blocking KHK Prevents and Reverses Alcoholic Liver Disease
In a 14-month chronic alcohol model, wild-type mice developed severe hepatic steatosis and inflammation, with triglyceride accumulation and upregulated proinflammatory cytokines (IL-6, Ccl2). In contrast, liver-specific KHK knockout mice were nearly completely protected, showing marked reductions in liver enzymes (AST, ALT) and fibrosis markers. This indicates that ethanol-induced liver injury depends on KHK-mediated fructose metabolism.
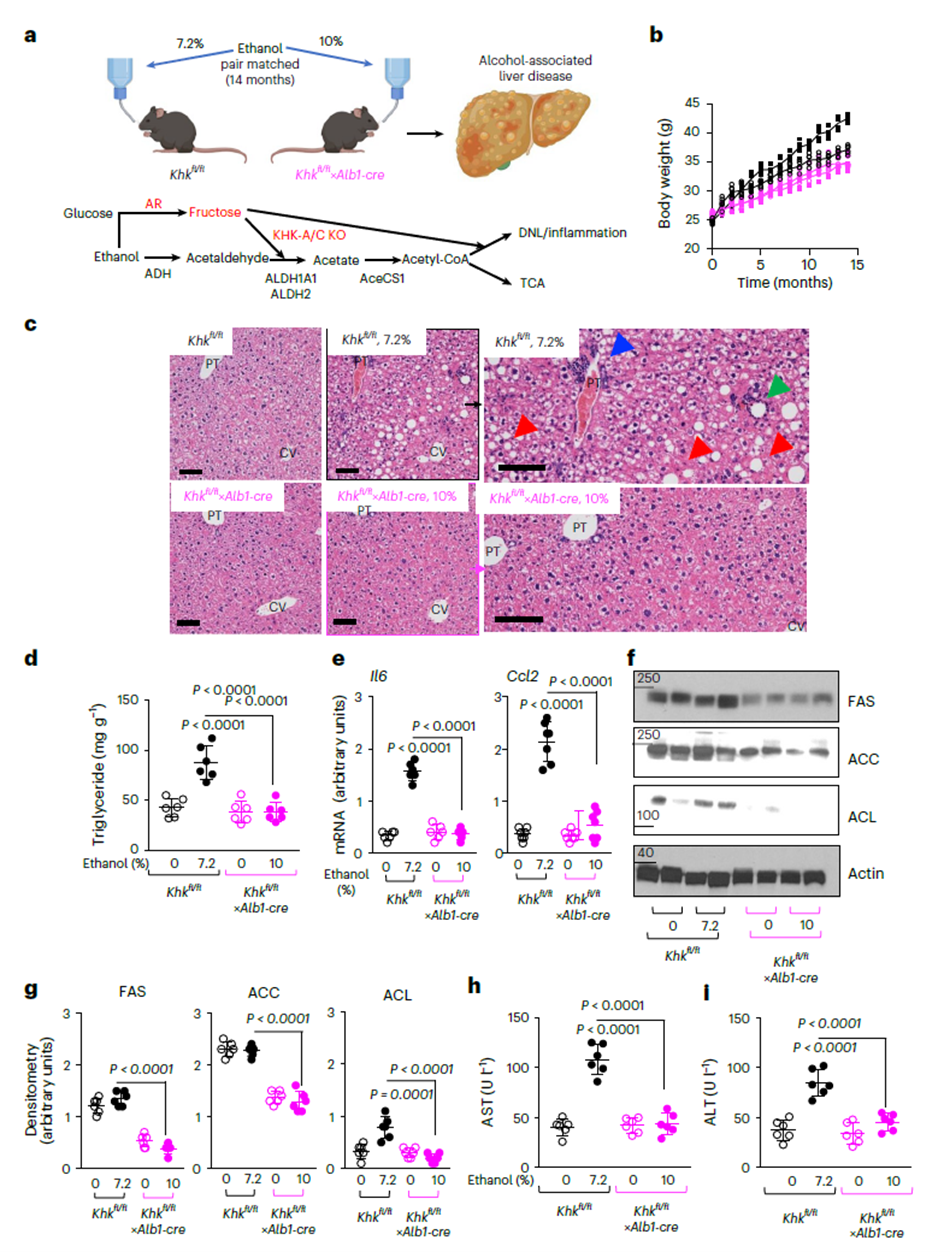
Figure 5. Blocking hepatic fructose metabolism protects against ALD
Moreover, an inducible KHK deletion model (Khkfl/fl×Ubc-cre) confirmed KHK’s reversible role: inducing KHK knockout in mice with established alcohol preference gradually reduced intake and preference, alongside downregulated ALDH and slowed acetaldehyde clearance. Pharmacologically, the KHK inhibitor CRP427 significantly reduced voluntary alcohol consumption in high-drinking models (cHAP mice and self-administering rats), validating KHK as a viable therapeutic target.
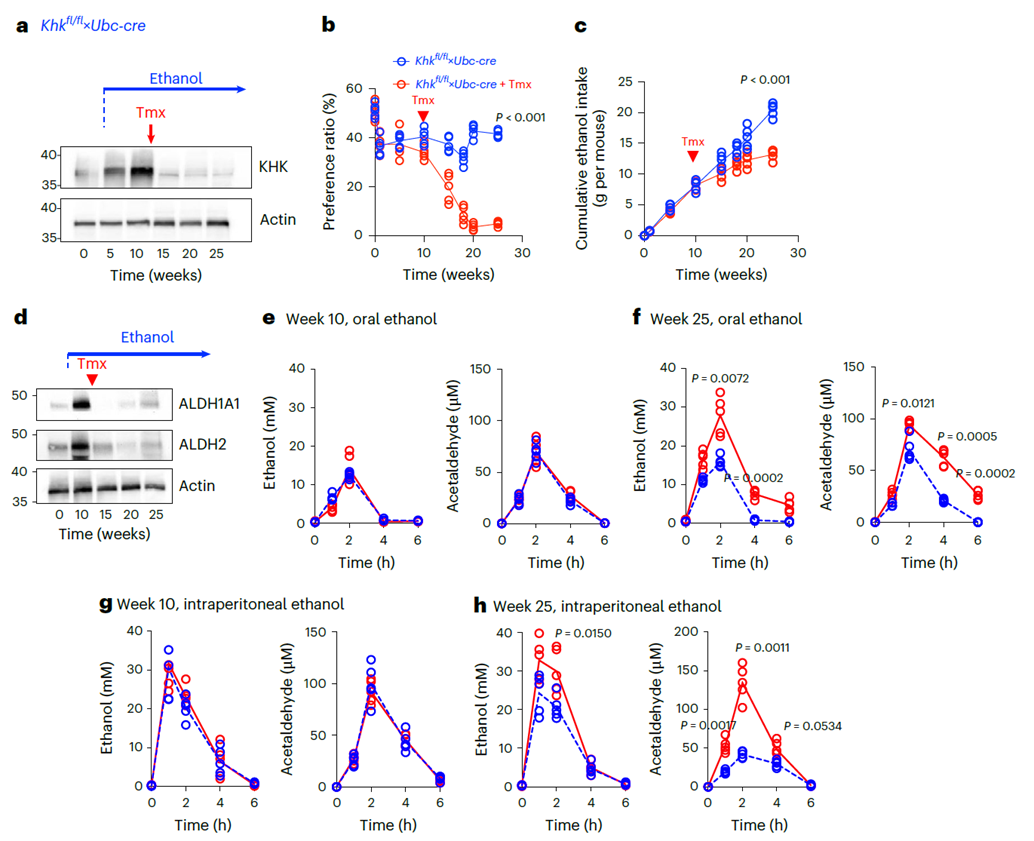
Figure 6. Targeted KHK-A/C gene knockout reverses ethanol preference and intake in mice
KHK is the rate-limiting enzyme in fructose metabolism, catalyzing the conversion of fructose to fructose-1-phosphate, providing substrate for lipid synthesis and energy storage. Its C isoform (KHK-C), predominantly expressed in the liver, is the primary driver of fructose-induced fat deposition and oxidative stress. Prior studies showed that KHK deletion significantly ameliorates high-fructose diet-induced non-alcoholic fatty liver and protects against metabolic syndrome and kidney injury. This study is the first to directly link KHK to alcohol preference and alcoholic liver disease (ALD), revealing its multifaceted roles in central reward, liver metabolism, and gut hormone regulation.
About abinScience
abinScience was founded in Strasbourg, France, leveraging the region’s world-class research ecosystem to focus on developing and producing high-quality life science reagents. Guided by the vision of “Empowering Bioscience Discovery,” abinScience is committed to providing efficient, reliable experimental solutions to researchers worldwide, accelerating cutting-edge life science research.
abinScience KHK-Related Proteins and Antibodies:
| Catalog No. |
Product name |
| HW836012 |
Recombinant Human KHK Protein, N-His |
| HW836014 |
Anti-Human KHK Polyclonal Antibody |
Disclaimer: This article is based on publicly available literature. Products are for research use only.

 中文
中文 English
English 한국어
한국어 日本語
日本語 Español
Español Français
Français Русский
Русский


