Neuropathic pain (NP) is a chronic pain state caused by peripheral nerve injury or diseases such as diabetes and cancer, imposing severe psychological and economic burdens on patients. Although its pathogenesis remains incompletely understood, persistent neuroinflammation and synaptic dysregulation in the spinal dorsal horn (SDH) have been established roles as key drivers. As the primary immune cells of the central nervous system, microglia play a central role in regulating neuroinflammation and synaptic pruning. However, how metabolic dysregulation in microglia—particularly lipid metabolism imbalance—affects their function and contributes to the onset and progression of NP remains poorly understood. This study focuses on the regulatory role of the transcription factor FOXA2 in microglial lipid metabolism. Using the spared nerve injury (SNI) mouse model and LPS-stimulated in vitro experiments, combined with techniques including immunofluorescence, transmission electron microscopy, qRT-PCR, Western blotting, and transcriptome sequencing, we demonstrate that FOXA2 regulates CPT1A expression through an HDAC3-mediated deacetylation mechanism, thereby restoring lipid metabolic homeostasis, alleviating neuroinflammation, and correcting synaptic pruning imbalance. This work highlights the therapeutic potential of targeting microglial lipid metabolism in NP and provides a theoretical foundation for developing novel pain management strategies.
FOXA2 Expression Is Significantly Downregulated in Neuropathic Pain Models
To establish the relationship between FOXA2 and NP, the research team first constructed a mouse NP model using spared nerve injury (SNI). SNI mice exhibited a marked reductions in paw withdrawal threshold (PWT) to mechanical stimuli and prolonged cold withdrawal latency, confirming successful model establishment. Western blot and qRT-PCR analyses revealed that both protein and mRNA levels of FOXA2 in the spinal dorsal horn were significantly reduced starting 4 days post-SNI and persisted until day 14. Immunofluorescence staining showed that FOXA2 primarily co-localized with the microglial marker Iba-1 and the neuronal marker NeuN, with a clear decrease in FOXA2 fluorescence intensity in microglia of SNI mice. In vitro, lipopolysaccharide (LPS) stimulation of primary microglia and BV2 cells similarly caused a marked downregulation of FOXA2 protein and mRNA levels, accompanied by reduced fluorescence intensity. These findings indicate that downregulation of FOXA2 may contribute to the pathological process of NP, laying the groundwork for subsequent mechanistic investigations.
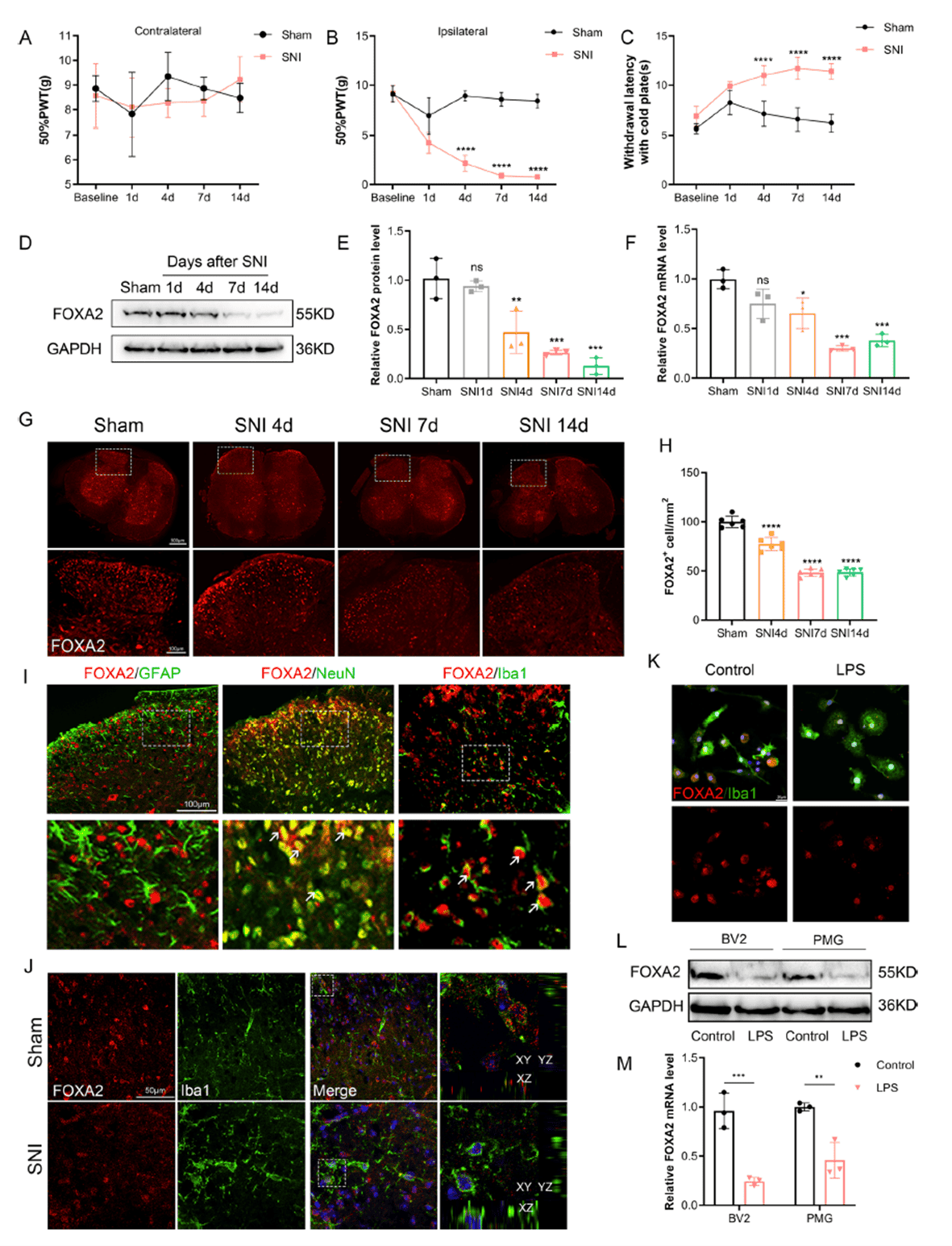
Figure 1. Decreased FOXA2 expression during neuropathic pain progression
FOXA2 Overexpression Significantly Alleviates Neuropathic Pain
To validate the functional role of FOXA2, the team achieved overexpression or knockdown of FOXA2 in the spinal dorsal horn via intrathecal injection of AAV vectors, with efficacy confirmed by Western blot and qRT-PCR. Behavioral tests showed that, compared with the control group (SNI+AAV-NC), FOXA2-overexpressing mice (SNI+AAV-Foxa2⁺) displayed significantly reduced mechanical and cold allodynia on the ipsilateral side, whereas FOXA2 knockdown exacerbated pain responses. No notable changes were observed on the contralateral side. Open-field test (OFT) results revealed no differences in total distance traveled or central zone entries among groups, indicating that AAV intervention did not affect locomotor activity and ruling out motor impairment as a confounding factor in pain assessment. These results clearly demonstrate the analgesic role of FOXA2 in NP, providing direct evidence for FOXA2-targeted therapeutic strategies.
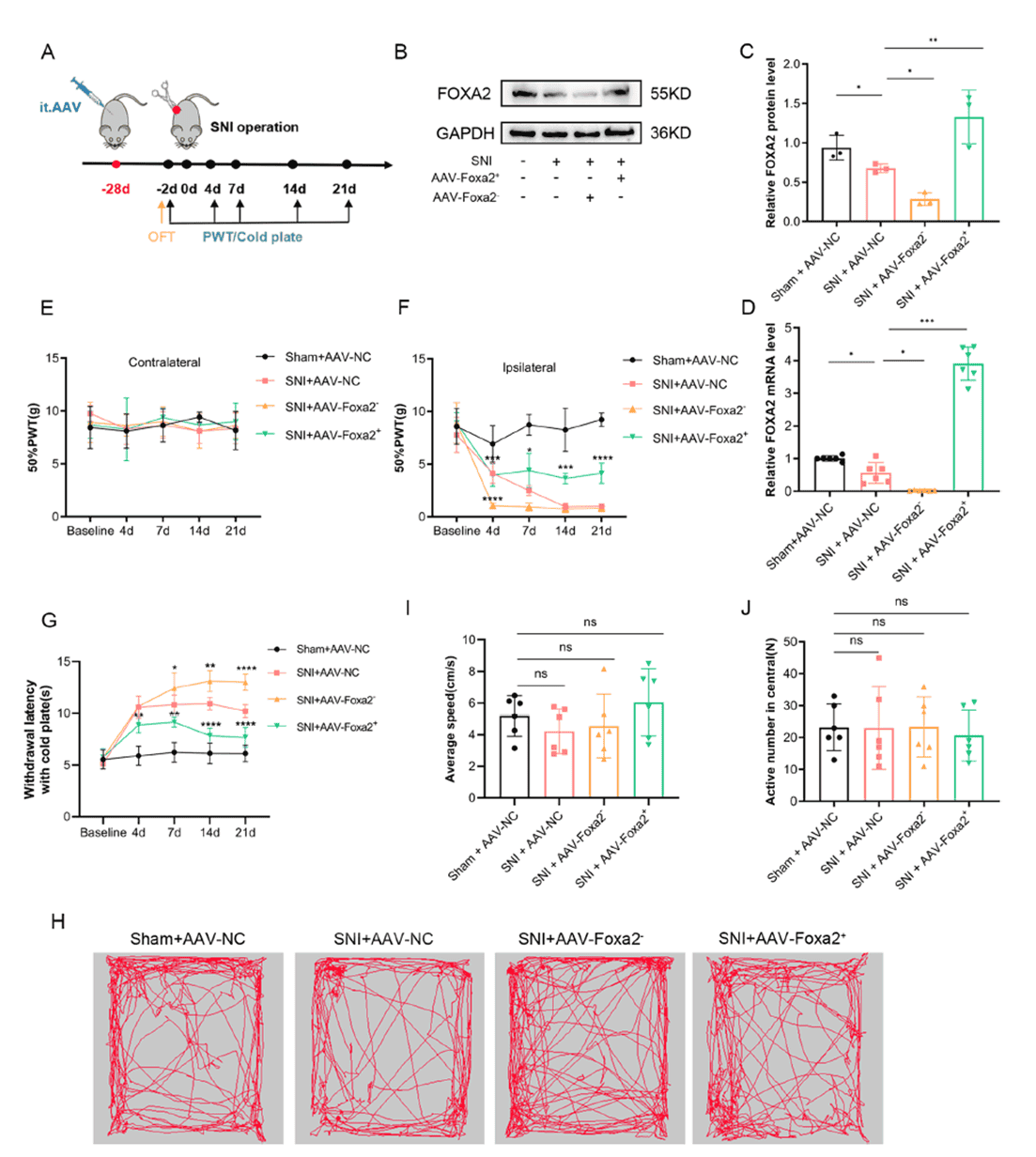
Figure 2. FOXA2 overexpression in the spinal cord induces analgesic effects
FOXA2 Reduces Microglial Oxidative Stress by Regulating Lipid Metabolism
To uncover the molecular mechanism underlying FOXA2’s effects, transcriptome sequencing was performed on spinal cord tissue from SNI+AAV-NC and SNI+AAV-Foxa2⁺ mice. GO and KEGG enrichment analyses revealed significant enrichment of differentially expressed genes in lipid metabolism-related pathways. BODIPY staining demonstrated that SNI triggered microglial activation and lipid droplet (LD) accumulation in the spinal dorsal horn, a phenomenon markedly reversed by FOXA2 overexpression. FOXA2 overexpression also reduced levels of the lipid peroxidation product MDA, suggesting attenuation of lipid toxicity-associated oxidative damage. In vitro, LPS stimulation dramatically increased lipid droplet numbers and reactive oxygen species (ROS) levels in primary microglia, effects that were effectively counteracted by FOXA2 overexpression (oeFoxa2) but exacerbated by FOXA2 knockdown (siFoxa2). Notably, the lipid droplet degradation inducer forskolin reversed the effects of FOXA2 knockdown. These data indicate that FOXA2 alleviates NP by maintaining microglial lipid metabolism homeostasis and reducing oxidative stress.
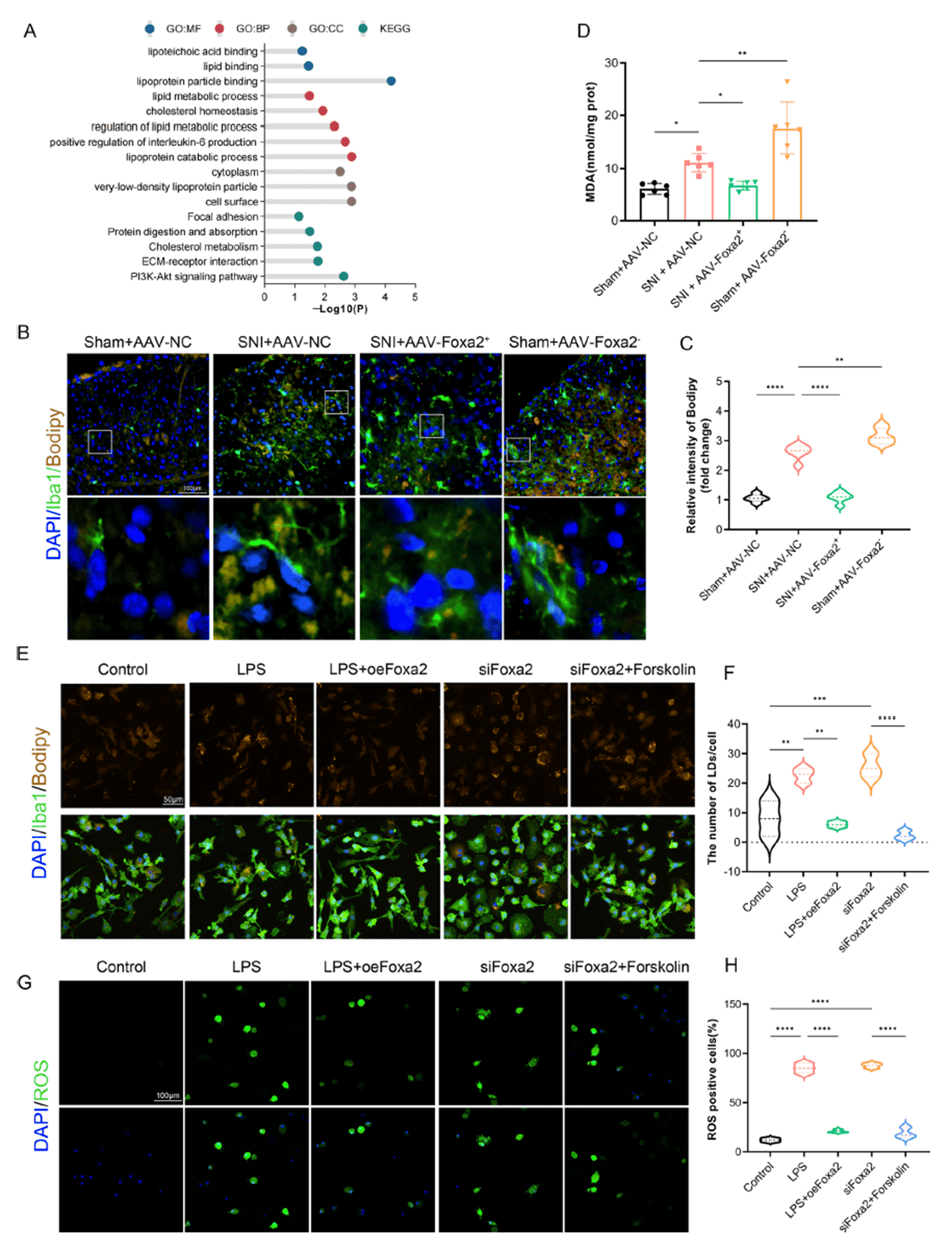
Figure 3. FOXA2 alleviates lipid toxicity and oxidative stress
FOXA2 Improves Microglial Mitochondrial Function and Energy Metabolism
Given that ROS primarily originate from mitochondrial electron transport, the team next investigated the impact of FOXA2 on mitochondrial function. Transmission electron microscopy (TEM) revealed swollen mitochondria with reduced cristae in SNI microglia, with FOXA2 knockdown causing even more severe swelling, fragmentation, and vacuolization. JC-1 staining showed decreased mitochondrial membrane potential in both SNI and FOXA2-knockdown conditions, which was restored by FOXA2 overexpression. In vitro, FOXA2 overexpression reduced LPS-induced mitochondrial ROS (mtROS), mitigated membrane potential depolarization, and restored ATP production in primary microglia, whereas FOXA2 knockdown mimicked LPS effects of LPS, inducing mitochondrial dysfunction that was partially rescued by forskolin. These results confirm that FOXA2 preserves mitochondrial structural and functional integrity, thereby supporting energy metabolism essential for anti-inflammatory and synaptic regulatory functions in microglia.
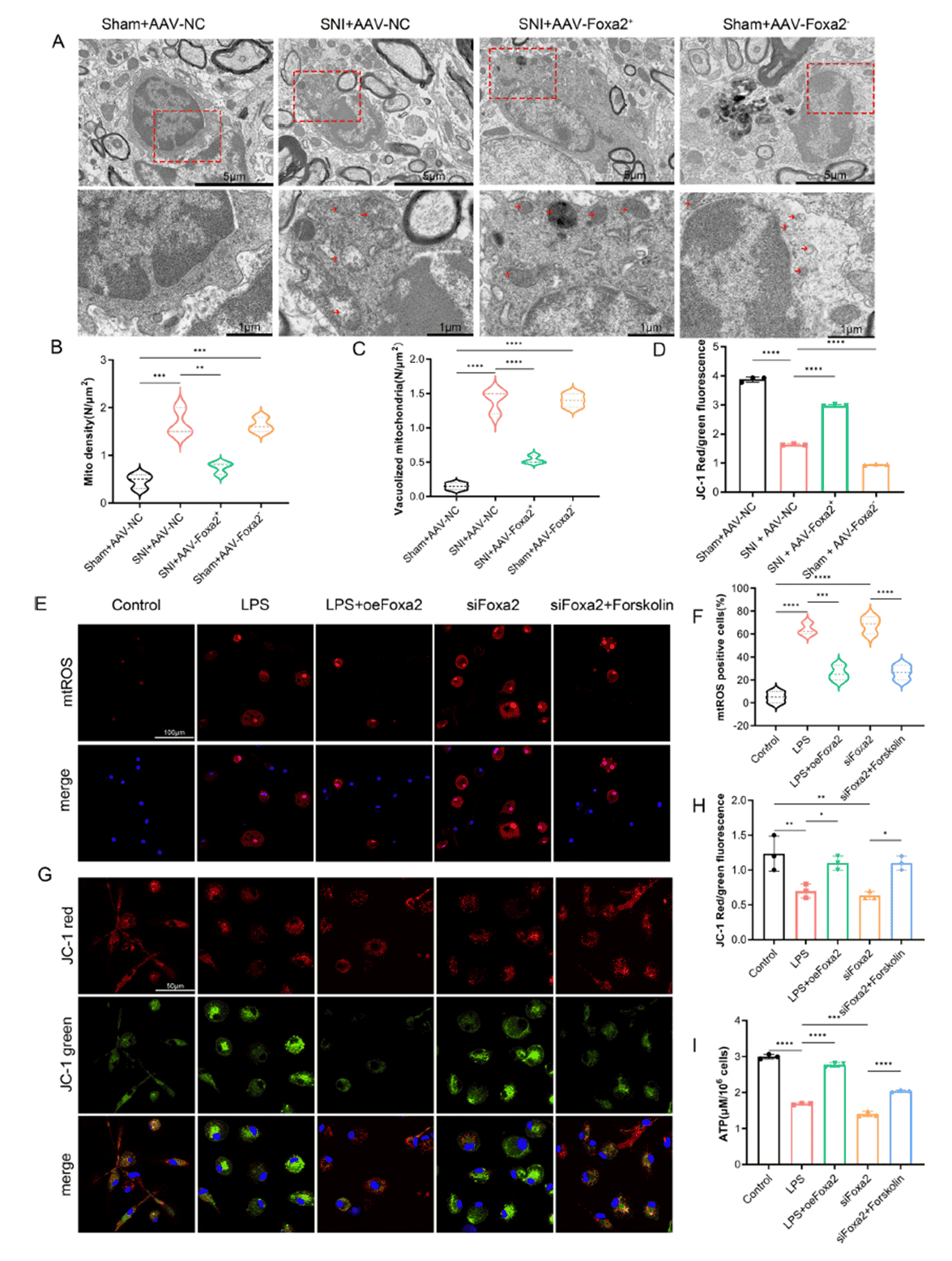
Figure 4. FOXA2 ablation leads to mitochondrial dysfunction and impaired energy metabolism
FOXA2 Promotes Anti-inflammatory Phenotype Switching in Microglia
Considering the close link between mitochondrial dysfunction and inflammatory responses, the team examined FOXA2’s influence on microglial polarization. Immunofluorescence showed that in SNI mice (SNI+AAV-NC), the proportion of pro-inflammatory CD86-positive microglia markedly increased while anti-inflammatory CD206-positive cells decreased in the spinal dorsal horn. FOXA2 overexpression (SNI+AAV-Foxa2⁺) reversed this shift, increasing CD206 and reducing CD86 expression. In vitro, LPS induced a similar pro-inflammatory skew that was corrected by oeFoxa2 but worsened by siFoxa2, with forskolin rescuing the siFoxa2 phenotype. These findings demonstrate that FOXA2 drives microglia toward an anti-inflammatory phenotype by regulating lipid metabolism, thereby attenuating neuroinflammation and contributing to NP relief.
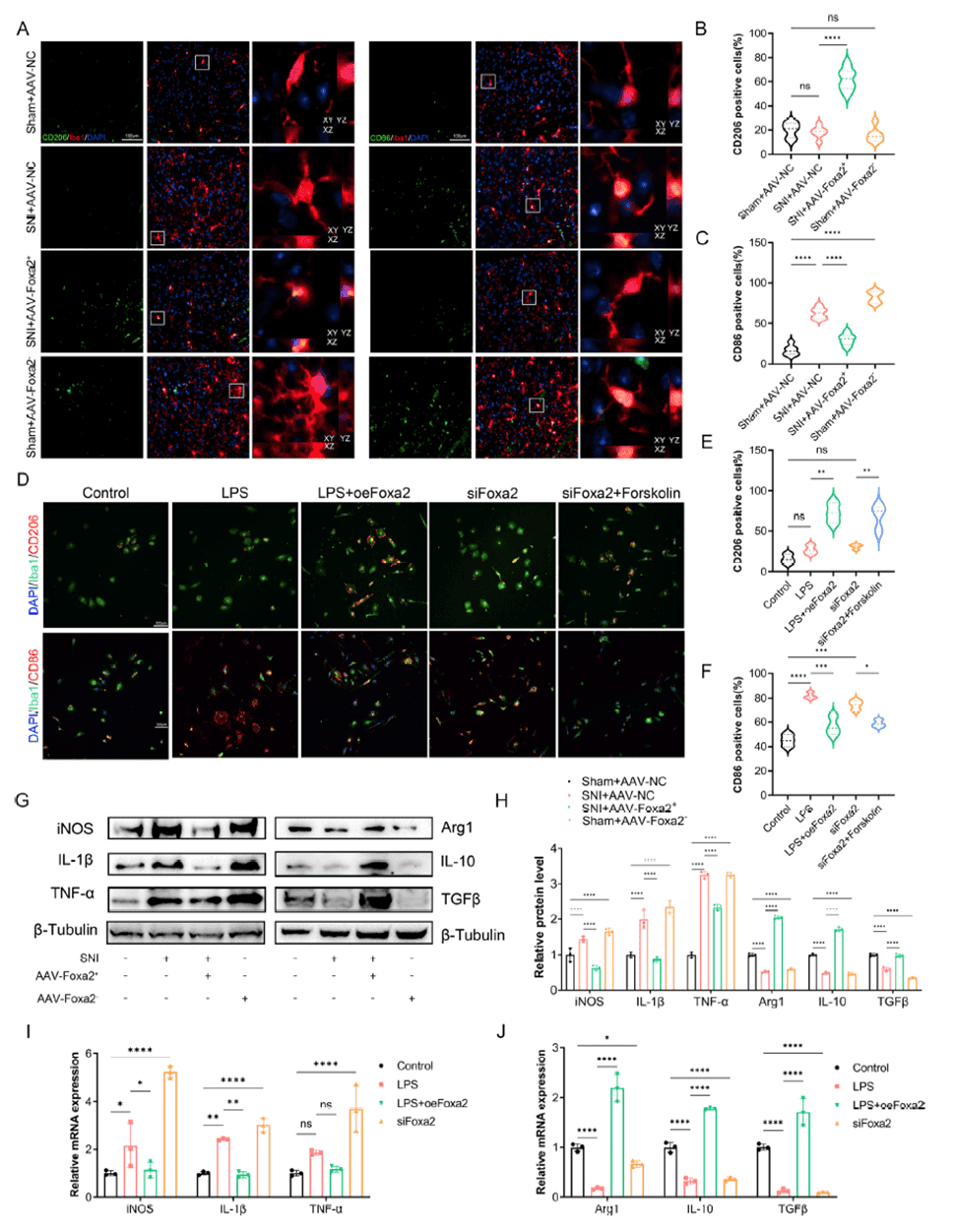
Figure 5. FOXA2 modulates inflammatory cytokine levels in microglia
FOXA2 Restores Microglial Synaptic Pruning and Corrects Synaptic Imbalance
A hallmark of neuropathic pain is excitatory/inhibitory (E/I) synaptic imbalance in the spinal dorsal horn, potentially driven by dysfunctional microglial synaptic pruning. Immunofluorescence co-localization analysis revealed that SNI significantly reduced the VGAT/Gephyrin (inhibitory synapse) ratio and increased VGLUT2/PSD95 (excitatory synapse) density in laminae I–II; FOXA2 overexpression ameliorated this imbalance, whereas knockdown worsened it. TEM showed that SNI and FOXA2 knockdown decreased symmetric (inhibitory, blue arrows) and increased asymmetric (excitatory, red arrows) synapses, with thickened postsynaptic densities and elongated active zones—changes reversed by FOXA2 overexpression. CD68 staining indicated reduced phagocytic activity in SNI and FOXA2-knockdown microglia, which was restored by FOXA2 overexpression. Moreover, FOXA2 overexpression enhanced phagocytosis of excitatory synapses while reducing ingestion of inhibitory synapses, with opposite effects upon knockdown. These results confirm that FOXA2 corrects E/I synaptic imbalance in the spinal dorsal horn by restoring microglial synaptic pruning, thereby alleviating central sensitization associated with NP.
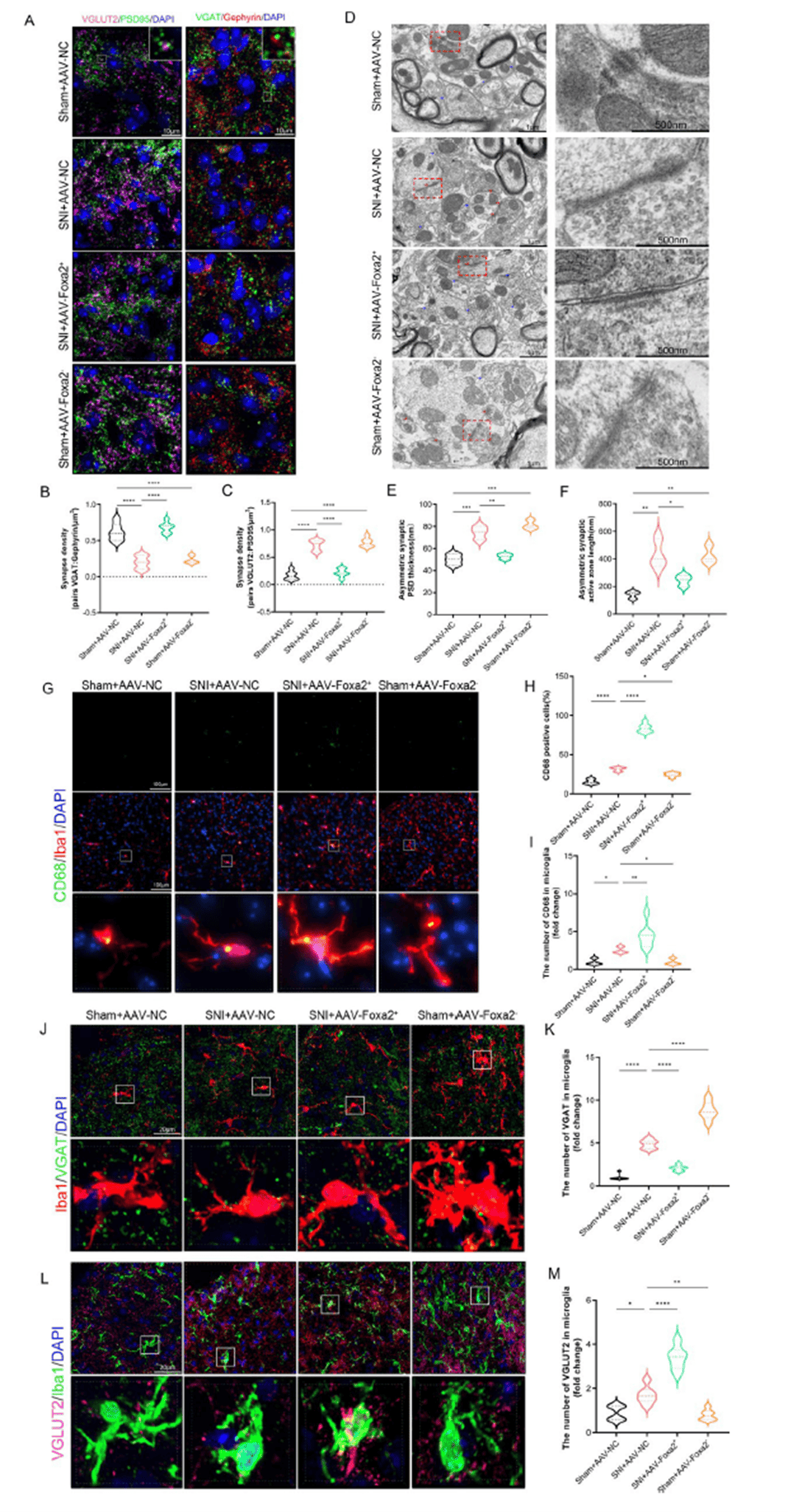
Figure 6. FOXA2 promotes microglia-mediated synaptic pruning
FOXA2 Regulates Lipid Metabolism and Synaptic Pruning via CPT1A
Transcriptome analysis revealed that FOXA2 overexpression significantly upregulated the lipid metabolism gene Cpt1a, with Western blot and qRT-PCR confirming a positive correlation between FOXA2 and CPT1A expression. Functional validation showed that Cpt1a knockdown abolished the analgesic effect of FOXA2 overexpression, blocked lipid droplet clearance, increased MDA levels, and exacerbated mitochondrial dysfunction. Synaptic analyses demonstrated that Cpt1a knockdown impaired FOXA2-mediated restoration of synaptic balance and reversed its effects on microglial phagocytosis preferences. These data establish CPT1A as a key downstream effector through which FOXA2 governs microglial lipid metabolism and synaptic pruning.
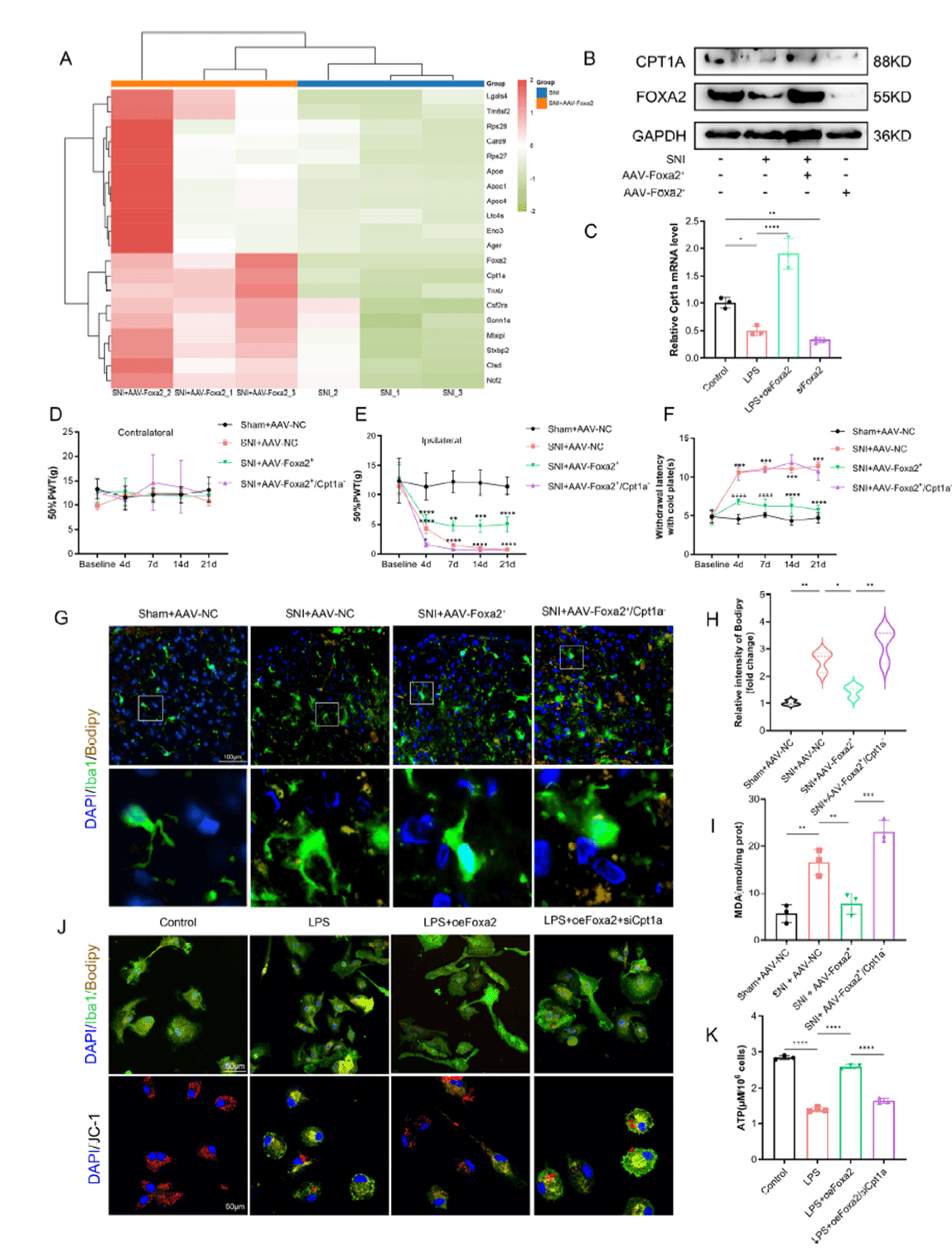
Figure 7. FOXA2 regulates microglial lipid metabolism and synaptic pruning through CPT1A
HDAC3 Inhibits FOXA2 Transcriptional Activity through Deacetylation
To identify the upstream regulator of FOXA2, the team screened selective inhibitors and found that the HDAC3-specific inhibitor RGFP966 dramatically upregulated FOXA2 expression in BV2 cells. Co-immunoprecipitation confirmed a direct interaction between HDAC3 and FOXA2, with RGFP966 increasing FOXA2 acetylation. Dual-luciferase reporter and ChIP-qPCR assays showed that FOXA2 transcriptionally activates Cpt1a by binding site 3 in its promoter, a promoter, a process inhibited by HDAC3 overexpression. In vivo, intrathecal RGFP966 administration significantly relieved pain behaviors in SNI mice, reduced lipid droplet accumulation in the spinal dorsal horn, suppressed pro-inflammatory cytokines, and enhanced anti-inflammatory markers. These results indicate that HDAC3 suppresses FOXA2 transcriptional activity via deacetylation, thereby modulating CPT1A expression and microglial function.
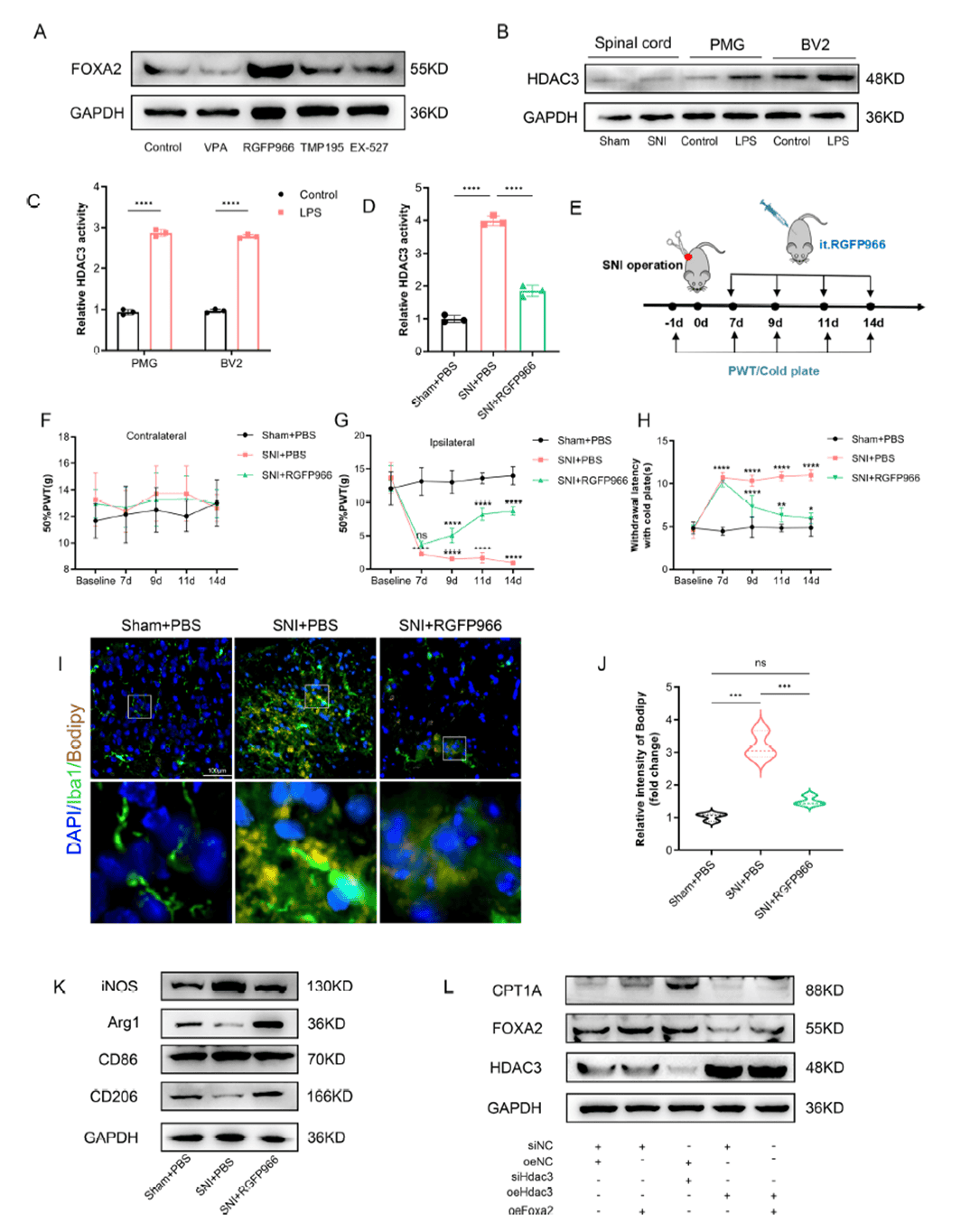
Figure 8. FOXA2 is regulated by HDAC3 activity and expression levels
This study, for the first time, elucidates the critical role of the HDAC3-FOXA2-CPT1A signaling axis in neuropathic pain. SNI or LPS stimulation increases HDAC3 activity in microglia, which deacetylates and inhibits FOXA2, leading to reduced CPT1A expression, lipid metabolism disorder, lipid droplet accumulation, mitochondrial dysfunction, exacerbated neuroinflammation, and impaired synaptic pruning—ultimately driving NP progression. Conversely, FOXA2 overexpression or HDAC3 inhibition restores CPT1A-mediated fatty acid oxidation, normalizes microglial metabolism, promotes anti-inflammatory polarization, and reinstates proper synaptic pruning, thereby alleviating neuropathic pain. This discovery not only establishes the central importance of microglial lipid metabolism in NP pathogenesis but also offers a promising new therapeutic target. Targeting the HDAC3-FOXA2-CPT1A axis to restore microglial metabolic and functional homeostasis represents a potential effective strategy for treating neuropathic pain.
abinScience provided the HRP-conjugated secondary antibody (TF690314) used in this study, serving as a key signal detection tool in Western blot experiments and providing reliable protein expression validation for elucidating the core mechanism of “HDAC3-FOXA2-CPT1A axis regulating microglial lipid metabolism, improving synaptic pruning disorder, and ultimately alleviating neuropathic pain.” abinScience was founded in Strasbourg, France, leveraging the region’s outstanding scientific innovation ecosystem to focus on the R&D and production of high-quality life science reagents. Guided by the vision “Empowering Bioscience Discovery,” abinScience is committed to delivering efficient and reliable experimental solutions to researchers worldwide, powering cutting-edge biomedical research.
Disclaimer: This article is a summary based on publicly available literature. All products mentioned are for research use only.

 中文
中文 English
English 한국어
한국어 日本語
日本語 Español
Español Français
Français Русский
Русский


